2B9R/cyclin B1構造/241112
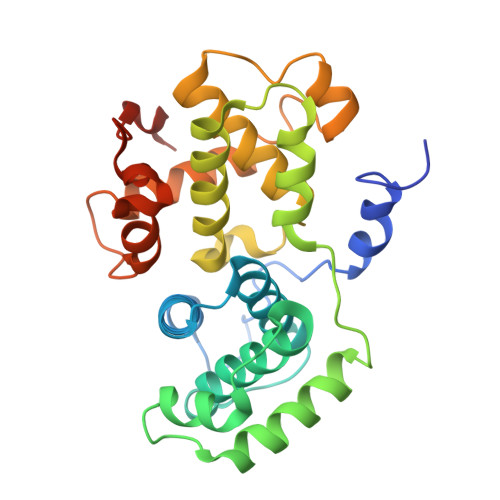
2B9R
Crystal Structure of Human Cyclin B1
- PDB DOI: https://doi.org/10.2210/pdb2B9R/pdb
- Classification: CELL CYCLE
- Organism(s): Homo sapiens
- Expression System: Escherichia coli
- Mutation(s): Yes
- Deposited: 2005-10-12 Released: 2006-10-17
Experimental Data Snapshot
- Method: X-RAY DIFFRACTION
- Resolution: 2.90 Å
- R-Value Free: 0.308
- R-Value Work: 0.243
- R-Value Observed: 0.249
PDBのwwPDB検証レポートのメトリクスは、タンパク質構造の信頼性と正確性を評価するための重要な指標です。以下に、主なメトリクスとその解釈方法を説明します:
R値(R-value): 実験データとモデルの一致度を示します。低いR値は、モデルが実験データに良く一致していることを示します。
Rフリー値(R-free value): モデルの過剰適合を防ぐために使用される指標で、モデルの信頼性を評価します。R値と同様に、低いRフリー値が望ましいです。
Ramachandranプロット: タンパク質の二次構造のジアヘドラル角(φとψ)の分布を示します。許容範囲内に多くの残基があるほど、モデルの品質が高いとされます。
RSRZ(Real Space R-value Z-score): 実空間でのモデルと実験データの一致度を示します。RSRZが2以下であれば、モデルは良好とされます。
幾何学的異常値(Geometric outliers): タンパク質の幾何学的パラメータ(結合長、結合角など)が標準からどれだけ外れているかを示します。異常値が少ないほど、モデルの品質が高いとされます。
これらのメトリクスを総合的に評価することで、タンパク質構造モデルの信頼性と正確性を判断することができます。詳細な情報は、wwPDBのFAQページやユーザーガイドで確認できます。
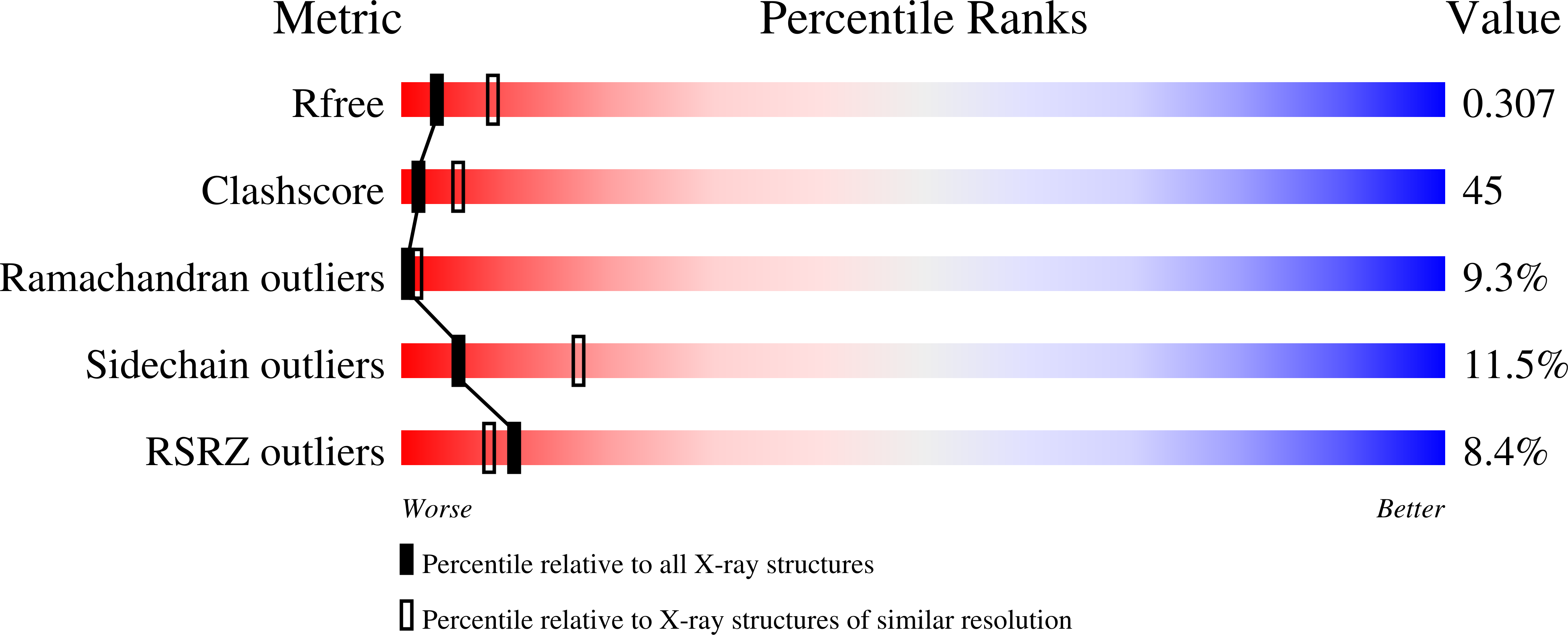
1. Overall quality at a glance
This section provides a succinct "executive" summary of key quality indicators. If there should be serious issues with a structure, this would usually be evident from this summary.
The metrics shown in the "slider" graphic (see examples below) compare several important global quality indicators for this structure with those of previously deposited PDB entries. The comparison is carried out by calculation of the percentile rank, i.e. the percentage of entries that are equal or poorer than this structure in terms of a quality indicator. The global percentile ranks (black vertical boxes) are calculated with respect to all X-ray structures available in the PDB archive up to 27 December 2023. The resolution-specific percentile ranks (white vertical boxes) are calculated with respect to a subset of X-ray entries in the same subset of the PDB archive, but only considering entries with comparable resolution to this entry. In general, one would of course like all sliders to lie far to the right in the blue areas (especially for recently determined structures, and especially the resolution-specific sliders).
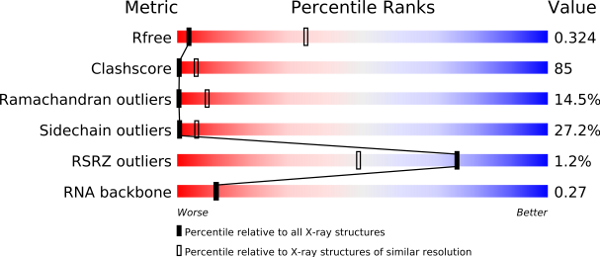
An example slider graphic for a relatively poor structure.

An example slider graphic for a relatively good structure.
Note that if you are not an expert you neither need to know what the various quality criteria measure nor whether the values for an entry are unusual or not. However, for increased understanding, below is a brief description of these key global quality indicators:
| Rfree | This is a measure of the fit of the model to a small subset of the experimental data, which was not used in model refinement (Brünger, 1992). The value quoted is from a recalculation by the DCC program (Yang et al., 2016). Further information can be found in the Data refinement and statistics section of the report, as described below. |
| Clashscore | This score is derived from the number of pairs of atoms in the model that are unusually close to each other. It is calculated by MolProbity (Chen et al., 2010) and expressed as the number or such clashes per thousand atoms. Further information can be found in the Close contacts section of the report, as described below. |
| Ramachandran outliers | A residue is considered to be a Ramachandran plot outlier if the combination of its φ and ψ torsion angles is unusual, as assessed by MolProbity (Chen et al., 2010). The Ramachandran outlier score for an entry is calculated as the percentage of Ramachandran outliers with respect to the total number of residues in the entry for which the outlier assessment is available. Further information can be found in the Torsion angles, Protein backbone section of the report, as described below. |
| Sidechain outliers | Protein sidechains mostly adopt certain (combinations of) preferred torsion angle values (called rotamers or rotameric conformers), much like their backbone torsion angles (as assessed in the Ramachandran analysis). MolProbity considers the sidechain conformation of a residue to be an outlier if its set of torsion angles is not similar to any preferred combination. The sidechain outlier score is calculated as the percentage of residues with an unusual sidechain conformation with respect to the total number of residues for which the assessment is available. |
| RSRZ outliers | The real-space R-value (RSR) is a measure of the quality of fit between a part of an atomic model (in this case, one residue) and the data in real space (Jones et al., 1991). The RSR Z-score (RSRZ) is a normalisation of RSR specific to a residue type and a resolution bin (Kleywegt et al., 2004). RSRZ is calculated only for standard amino acids and nucleotides in protein, DNA and RNA chains. A residue is considered an RSRZ outlier if its RSRZ value is greater than 2. The RSRZ outlier score as shown in the slider graph is calculated as the percentage RSRZ outliers with respect to the total number of residues for which RSRZ was computed. This is calculated by the EDS (Electron-Density Server) component of the validation pipeline. |
| RNA backbone | Like the protein backbone and sidechains, the RNA backbone also adopts certain sets of preferred torsion angle values. Based on statistical analysis of RNA chains in the PDB, MolProbity (Chen et al., 2010) assigns a score per nucleotide for the quality of its backbone. This metric is calculated as the average score of all nucleotides in the entry. |
| For more information about validation metrics, see Read et al. (2011) and the review by Kleywegt (2000). |
The slider graph is followed by a table that shows the number of entries upon which the percentile rank calculations are based, and the resolution ranges in which they lie:
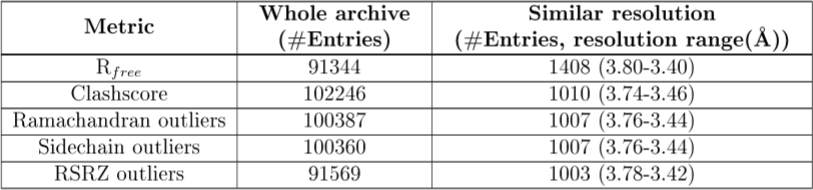
| The resolution-specific ranks are calculated using the smallest bin width that includes at least 1,000 entries. The resolution used is the highest resolution of data used in the refinement. |
The next table provides a graphical summary of the quality of all polymeric chains:
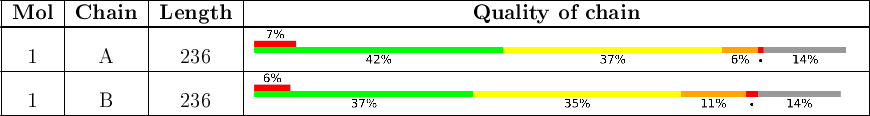
There may be green, yellow, orange and red portions in the lower bar for each chain, indicating the fraction of residues that contain outliers for 0, 1, 2, ≥3 model-only validation criteria, respectively. A grey segment indicates residues present in the sample but not modelled in the final structure. If electron density outliers were present, there is an additional red bar above the lower bar, indicating the fraction of residues that are RSRZ outliers. The numeric value for each fraction is shown below the corresponding segment. Values <5% are indicated with a dot. If electron density outliers were present, there is an additional red bar above the lower bar, indicating the fraction of residues that are RSRZ outliers. The numeric value for the fraction is indicated above this red bar.
| The Quality of chain chart shows the fraction of residues in each chain that are flagged as unusual according to the validation criteria used rather than where in the sequence this occurs (the plots are a kind of horizontal pie chart). The following section Residue-property plots provides a graphic showing where in the sequence the issues occur. |
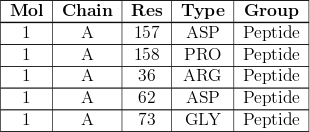
If the structure contains ligands, carbohydrates or non-standard residues that have outliers for geometric and/or electron-density-fit criterion, an additional table will be presented:

This provides a summary of which residues have issues. The columns are labelled:
| Mol | The identifier of the molecule (for experts: this is the same as the "entity id" in the mmCIF file of the entry) |
| Type | The residue name as defined in the PDB chemical component dictionary |
| Chain | The instance identifier |
| Res | The residue number. Where applicable, an insertion code and alternative conformation identifier are specified as well. |
| Chirality | an X mark indicates there is some problem found with a chiral atom in the molecule, further details can be found in the Model quality section of the report as described below |
| Geometry | an X mark indicates that more than 40% of bonds, angles, torsions, chiral centres and rings are found to be outliers. Further information on the specific issues can be found in the Model quality section of the report, as described below. |
| Clashes | an X mark indicates that the molecule is involved in some close interatomic contacts, further details can be found in the Close contacts section of the report as described below |
| Electron density | an X mark is used where the ligand residue has Real-Space R-value (RSR) exceeding 0.3 and a Real-Space Correlation Coefficient (RSCC) that is less than 0.8 so that its fit to the EDS-style electron density maps may well indicate that "the experimental data do not accord with the ligand placement" (Smart et al., 2018). Further details can be found in the Fit of model and data section of the report, as described below. |
2. Entry composition
This section summarises the number of unique molecules that are present in the entry, and how they have been modelled. Each unique molecule and its instances (chain id) are described in a table:

with the following columns:
| Mol | The identifier of the molecule (for experts: this is the same as the "entity id" in the mmCIF file of the entry). |
| Chain | The instance identifier. If there is more than one model present in the entry, the chain is prefixed with a model number. |
| Residues | The number of residues in the molecule. |
| Atoms | This tabulates the counts of various element types in the molecule. |
| ZeroOcc | The number of atoms modelled with zero occupancy in the molecule. |
| AltConf | The number of residues that have been modelled with at least one alternative conformation. |
| Trace | The number of residues in the molecule that have been modelled with a reduced set of atoms. Protein or nucleic acid chains may be modelled with only one or two atoms (e.g. Cα, Cβ, P, an atom in a sugar ring or nucleobase, etc.). Typically, such cases are observed when the resolution is insufficient to confidently model all atoms. |
In addition, each unique oligosaccharide molecule, if present, is represented with a 2D SNFG image (Tsuchiya et al., 2017).
A Ligand Of Interest (LOI) is a subject of author’s research. Ligands that are flagged by authors during deposition are labelled as LOI.
| The Mol and Chain identifiers are also used in other tables in the report. |
3. Residue-property plots
This section shows summary plots of quality information for protein, RNA and DNA molecules on a per-residue basis.
There are two graphics shown for each molecule. The first graphic is the same as that shown in section 1: the green, yellow, orange and red segments indicate the fraction of residues with 0, 1, 2 and 3 or more types of model-only quality criteria with outliers, respectively. The additional red segment above the summary graphic (if present) indicates the fraction of residues that have an unusual fit to the density (RSRZ outliers).
The second graphic shows the sequence annotated by these criteria with outliers in model quality and unusual fit to the electron density (see example graphic below). The colour-coding described above is used here too. A red dot above a residue indicates a poor fit to the electron density (i.e., an RSRZ outlier). Consecutive stretches of residues for which no outliers were detected at all are not shown individually, but indicated by a green connector. Residues absent from the final model are shown in grey.
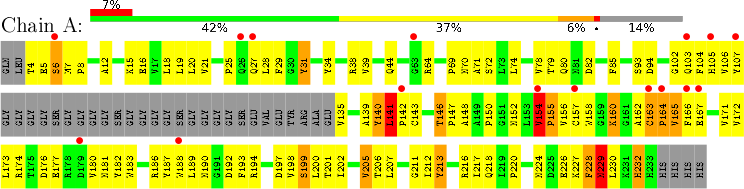
In general, the less red, orange, yellow and grey these plots contain, the better. It is important to realise that residues that are outliers on one or more model-validation criteria could be either errors in the model, or reflect genuine features of the structure. Careful analysis of the experimental data (electron density maps) is typically required to make the distinction. Outlier residues that are important for structure or function (e.g., enzymatic residues, interface residues, ligand-binding residues) should be inspected extra carefully (and addressed in a manuscript describing the structure).
The types of model-only quality criteria included in this analysis, and the software used for their calculation are:
Details of the outliers found for a residue can be found further down the report, in the Model quality section. |
4. Data and refinement statistics
This section provides a summary of the data and refinement statistics for the entry, including the source of the information. The precise definitions of many of the metrics can be found in original publications and crystallography textbooks. The following information is included in the table:
| Space group | This is the symmetry system in which the sample was crystallised to obtain the diffraction data. |
| Cell constants | These are the unit-cell dimensions and angles. |
| Data completeness | The number of expected diffraction spots is a function of data resolution and the space group. This metric describes the number of recorded reflections as a percentage of the number expected. It is reported both as provided by the depositor and as calculated by Servalcat (command refmacat). |
| Rmerge, Rsym | These metrics give an indication of the agreement between multiple intensity measurements. |
| <I/σ(I)> | Each reflection has an intensity (I) and an uncertainty in measurement (σ(I)), so I/σ(I) is the signal-to-noise ratio. This ratio decreases at higher resolution. <I/σ(I)> is the mean of individual I/σ(I) values. Value for outer resolution shell is given in parentheses. In case structure factor amplitudes are deposited, Xtriage estimates the intensities first and then calculates this metric. When intensities are available in the deposited file, these are converted to amplitudes and then back to intensity estimate before calculating the metric. |
| Resolution | Lower and upper limits of the resolution of the diffraction data, as reported by the depositor (usually, the range of data used in refinement of the model) and as calculated by EDS (the range of data encountered in the deposited reflection file). |
| Refinement program | The software that the depositor used for the final crystallographic refinement. |
| R, Rfree | These metrics, calculated by the DCC program (Yang et al., 2016), measure the similarity between the observed structure-factor amplitudes and those calculated from the model. Lower values and smaller differences between the two statistics are usually better. Rfree should be higher than R because it is calculated using reflections not used in the refinement. |
| Wilson B-value | An estimate of the overall B-value of the structure, calculated from the diffraction data. It serves as an indicator of the degree of order in the crystal and the value is usually not hugely different from the average B-value calculated from the model. |
| Anisotropy | The ratio (Bmax ‑ Bmin) / Bmean where Bmax, Bmin and Bmean are computed from the B-values associated with the principal axes of the anisotropic thermal ellipsoid. This ratio is usually less than 0.5; for only 1% of PDB entries it is more than 1.0 (Read et al., 2011). |
| Bulk-solvent parameters | Disordered solvent occupies a significant fraction of the crystal and contributes to low-resolution reflection terms. Bsol and ksol are parameters in a formulation for estimation of bulk solvent contributions to structure factors, and they are estimated during refinement. |
| Estimated twinning fraction | Twinning is the phenomenon where at least two domains occur within a single crystal related by mathematical operations called twinning operators. The twinning fraction is estimated using the H-test (Yeates, 1988). The estimated fraction is listed along with the associated twinning operator. Twinning is not possible in all space groups. |
| L-test for twinning | This test, based on acentric reflections, defines two metrics < |L| > and < L2 >, whose theoretical values are 0.5 and 0.375 in the untwinned case, and 0.333 and 0.2 in the perfectly twinned case (Padilla & Yeates, 2003). |
| Outliers | These are reflections in the diffraction data with very unlikely magnitude for amplitude or intensities. These are identified by Xtriage using its "extreme value statistics" method on the largest normalised intensities (Read, 1999). More than 0.1% outliers in a dataset generally indicates a fundamental problem with the data, unless translational NCS is present. Xtriage’s assessment of possible translational NCS is printed below the table. |
| Fo,Fc correlation | The difference between the observed structure factors (Fo) and the calculated structure factors (Fc) measures the correlation between the model and the experimental data. This metric is calculated by Servalcat (command refmacat). |
| Total number of atoms | This is the total number of atoms modelled in the entry. |
| Average B, all atoms | This is the mean B-value calculated over all modelled atoms. |
5. Model quality
Quality statistics in this section are calculated using standard compilations of covalent geometry parameters (Engh & Huber, 2001; Parkinson et al., 1996), tools in MolProbity (Chen et al., 2010), Validation-pack (Feng et al.) and the wwPDB chemical component dictionary (CCD).
5.1. Standard geometry
This section describes the quality of the covalent geometry for protein, DNA and RNA molecules in terms of bond lengths, bond angles, chirality and planarity. There are two tables providing a per-molecule summary and four tables that provide information on (some of) the outliers for each criterion (if any; otherwise the table is omitted).
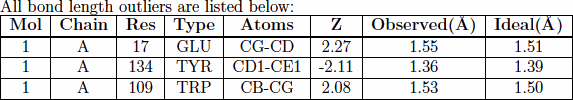
Summary table for bond lengths and angles
Expected bond length and bond angle values (and standard deviations) for standard amino acids and nucleotides are available in a wwPDB compilation (wwPDB, 2012). The MolProbity Dangle program calculates Z-scores of bond length and bond angle values for each residue in the molecule relative to the expected values. (A Z score is generally defined as the difference between an observed value an expected or average value, divided by the standard deviations of the latter.)
The root-mean-square value of the Z-scores (RMSZ) of bond lengths (or angles) is calculated for individual residues and then averaged for each chain and over the whole molecule. RMSZ scores are expected to lie between 0 and 1. For low-resolution structures, geometry should be tightly restrained and small values are expected. For very high-resolution structures, values approaching 1 may be attained. Values greater than 1 indicate over-fitting i of the data. Individual bond lengths or angles with a Z-score greater than 5 or less than -5 merit inspection.
The bond/angle summary table:
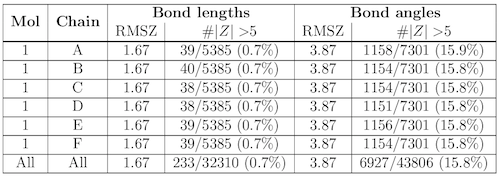
has the following columns:Mol The molecule identifier
Chain The instance identifier.
Bond lengths The RMSZ sub-column gives the Root Mean Squared Z score of all bond lengths analyzed.
The #|Z| >5 sub-column provides the number of bond lengths that have a Z-score > 5 or < -5 in comparison to the total number of bonds analyzed. †
Bond angles The RMSZ sub-column gives the Root Mean Squared Z score of all bond angles analyzed.
The #|Z| >5 sub-column provides the number of bond angles that have a Z-score > 5 or < -5 in comparison to the total number of angles analyzed. † :: † The percentage of outliers is listed in parentheses.
Deviations from expected chirality and planarity in the model are calculated by Validation-pack (Feng et al.).
Chiral centres for all compounds occurring in the PDB are described in the chemical component dictionary. Chirality can be assessed in a number of ways, including calculation of the chiral volume, e.g. for the Cα of amino acids this is 2.6 or -2.6 Å3 for L or D configurations, respectively. If the sign of the computed volume is incorrect, the handedness is wrong. If the absolute volume is less than 0.7Å3 , the chiral centre has been modelled as a planar moiety which is very likely to be erroneous. Chirality deviations are summarised per chain.
Three kinds of potential planarity deviations are assessed:
Sidechain: Certain groups of atoms in protein sidechains and nucleotide bases are expected to be in the same plane. An atom"s deviation from planarity is calculated by fitting a plane through these atoms and then calculating distance of individual atom from the plane. Expected value of such distances have been pre-calculated from data analysis (wwPDB, 2012). If an atom is modelled to be more than six times farther than the pre-calculated value, the residue is flagged to have a sidechain planarity deviation.
Peptide: A deviation is flagged if the omega torsion angle of a peptide group differs by more than 30° from the values expected for a proper cis or trans conformation (0° and 180°, respectively).
Main chain: The N atom of an amino acid residue is expected to be in the same plane as the Cα, C, and O atoms of the previous residue. If it is out of plane by more than 10°, this is flagged as a planarity deviation.
Outlier listing detailed tables
Where outliers exist, up to five for each category are listed in a table in the Summary Report, whereas the Full Report will list all the outliers found. For bond lengths and bond angles, the worst outliers are reported.
All the different outlier tables have the following columns in common:
| Mol | The molecule identifier. |
| Chain | The instance identifier |
| Res | The residue number. Where applicable, an insertion code and alternative conformation identifier are specified as well. |
| Type | The residue name. |
The following columns are specific to the bond length and bond angle outlier tables:
| Atoms | names of atoms involved in the bond or angle. |
| Z | The Z-score of the bond length or angle. |
| Observed | The observed value of the bond length or angle. |
| Ideal | The ideal value of the bond length or angle. |
For example: |
The following column is specific to the chirality outliers table:
| Atom | The name of the atom that is asssessed to have an unusual chiralty (see above for details of chirality assessment) |
For example: |

The following column is specific to the planarity outliers table:
| Group | The planarity deviation type, i.e. sidechain, main chain or peptide as described above. |
For example: |
5.2. Too-close contacts
This section provides details about too-close contacts between pairs of atoms that are not bonded where there is an unfavorable steric overlaps of van der Waals shells (clashes). Two kinds of close-contacts are analysed: those that occur between atoms in the same asymmetric unit (ASU) and those that occur between atoms in different ASUs. The latter are called symmetry-related clashes.
All-atom contacts within the ASU are calculated by the Reduce and Probe programs within MolProbity (Word et al., 1999; Chen et al., 2010). This method was developed to quantify the detailed non-covalent fit of atomic interactions within or between molecules (H-bonds, favorable van der Waals, and steric clashes). Since most such interactions involve H atoms on one or both sides, all hydrogens must be present or added (Reduce optimizes rotation of OH, SH, NH3, etc. within H-bond networks, but methyls stay staggered). At present, in order to ensure comparable scores between NMR and X-ray, hydrogen atoms are removed from the analysed structure, and replaced by a different set placed by Reduce in idealised and optimized nuclear-H positions. All-atom unfavorable overlaps ≥0.4Å are then identified as clashes, using van der Waals radii tuned for the nuclear H positions suitable for NMR (rather than the electron-cloud H positions suitable for X-ray). MolProbity then calculates an all-atom clashscore, which is defined as the number of clashes per 1000 atoms (including hydrogens). Percentile scores of the clashscore are also computed, to allow assessment of how the structure compares to the rest of the archive.
Symmetry-related clashes are identified by Validation-pack (Feng et al.). For each clash, a symmetry and translation code of the ASU of the clashing atom is also reported.
Clashes are summarised in a table, for example:
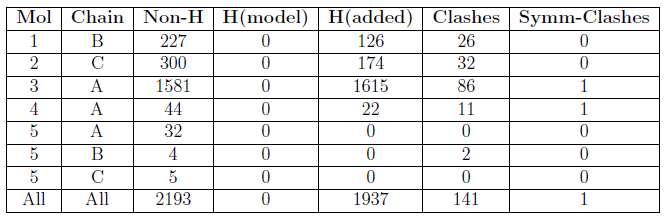
The columns are labelled:
| Mol | The molecule identifier |
| Chain | The instance identifier |
| Non-H | The number of non-hydrogen atoms modelled. |
| H(model) | The number of hydrogen atoms modelled. |
| H(added) | The number of hydrogen atoms added by MolProbity. |
| Clashes | The number of clashes in which the atoms in this instance of the molecule are involved. |
| Symm-clashes | The number of symmetry-related clashes in which the atoms in this instance of the molecule are involved. |
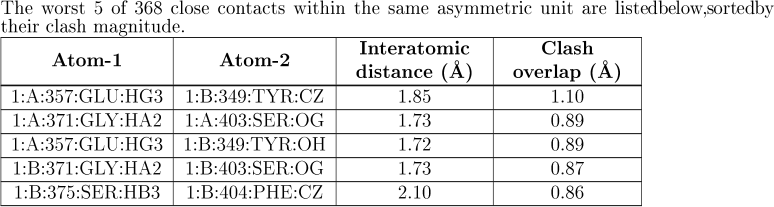
If there are clashes within the ASU a table with details will then be given:
 ]
]
the table has the following columns:
| Atom-1 | The molecule identifier, instance identifier, residue number, residue name and atom name for the first atom. where applicable, the chain identifier is prefixed with model number and an alternative conformation identifier is shown as a suffix to the atom name. |
| Atom-2 | Identifies the second atom in the clash. |
| Interatomic distance | The distance between Atom-1 and Atom-2 in Å. |
| Clash overlap | for within the asu clashes, the "magnitude" of the clash is assessed by MolProbity. the MolProbity "magnitude" of a clash is defined as the difference between the observed interatomic distance and the sum of the van der Waals radii of the atoms involved (Chen et al., 2010). The radii used are tuned for use with nuclear H positions suited for NMR (rather than the electron-cloud H positions used for X-ray). |
| In a Summary Report up to five of the worst clashes are listed in the table, whereas in a Full Report all the clashes are listed. |
If there are clashes to symmetry related atoms then a further table will be given:
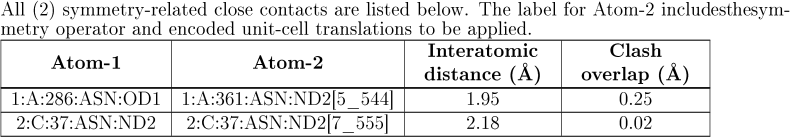
The table has the following columns:
| Atom-1 | Identifies the second atom in the clash. |
| Atom-2 | Identifies the second atom in the clash and the symmetry operator involved. |
| Interatomic distance | The distance between Atom-1 and Atom-2 in Å. |
| Clash overlap | For symmetry-related clashes, the criterion for identifying clashes is different. In this case, the "magnitude" of a clash is defined as 2.2 Å (or 1.6 Å if either atom is a hydrogen) minus the interatomic distance. |
| The 1_555 notation is crystallographic shorthand to describe a particular symmetry operator (the number before the underscore) and any required translation (the three numbers following the underscore). Symmetry operators are defined by the space group and the translations are given for the three-unit cell axis (a, b, and c) where 5 indicates no translation and numbers higher or lower signify the number of unit cell translations in the positive or negative direction. For example, 4_565 indicates the use of symmetry operator 4 followed by a one-unit cell translation in the positive b direction. For further information see RCSB PDB-101: Introduction to Biological Assemblies and the PDB Archive |
5.3. Torsion angles
5.3.1. Protein backbone
This section is populated if there are protein molecules present in the entry. The conformation of a protein backbone can be described by a pair of torsion angles (phi, psi) per residue (the remaining torsion angle, omega, is usually 180°). Ramachandran plots show the combinations of phi-psi values in a structure and typically compare these to a distribution of commonly observed values in high-resolution crystal structures. MolProbity’s Ramachandran plots are residue-type specific, derived from a high-quality subset of protein X-ray structures and divided into favoured, allowed and outlier regions. Favoured and allowed regions are defined to be the regions that include 98% and 99.95%, respectively, of the residues in the high-quality data (see (Chen et al., 2010). for more details).
This section contains a summary of analysis of the backbone torsion angles phi and psi by Molprobilty.
The summary table contains the following columns:
| Mol | The molecule identifier |
| Chain | The instance identifier |
| Analysed | The first number here is the number of residues in the chain for which MolProbity output is available. The second number is the total number of residues in the chain. Phi and psi angles cannot be analysed for terminal residues, non-standard residues or for residues with incompletely modelled main chain. |
| Favoured, Allowed, Outliers | The number (and percentage) of residues in the favoured, allowed and outlier regions respectively, of the residue-specific phi-psi plots. |
| Percentiles | The percentile score based on the percentage of Ramachandran outliers in the chain. These are given relative to the whole archive (first value) and relative to structures of a similar resolution (second value). The colours around the percentile values correspond to the slider positions in the Overall quality section of the report, as described above |
Where Ramachandran outliers exist, up to five randomly chosen outlier residues are listed in a table in the Summary Report, whereas the Full Report will list all the outliers found. It has following columns:
| Mol | The molecule identifier |
| Chain | The instance identifier |
| Res | The residue number |
| Type | The residue name |
5.3.2. Protein sidechains
Protein sidechain conformation can be described by the chi torsion angles. Depending on residue type, these angles adopt certain preferred sets of values (also termed rotamers or rotameric conformers). Based on analysis of high quality X-ray entries in the PDB, MolProbity assesses whether a sidechain is similar to one of the preferred sets of torsion angles, or is an outlier (see (Chen et al., 2010). for more details). This section is based on MolProbity analysis of sidechains.
The summary table summarises of sidechain outliers and has the following columns:
| Mol | The molecule identifier |
| Chain | The instance identifier |
| Analysed | The first number here is the number of residues in the chain which were analysed by MolProbity. The second number is the total number of residues in the chain. Chi torsion angles cannot be analysed for non-standard residues or for residues with incompletely modelled sidechains. |
| Rotameric, Outliers | The number (and percentage) of residues with favoured, and unusual chi torsion angles respectively. |
| Percentiles | The absolute and relative percentile scores based on the percentage of sidechain outliers in the chain. These are given relative to the whole archive (first value) and relative to structures of a similar resolution (second value). The colours around the percentile values correspond to the slider positions in the Overall quality section of the report, as described above |
Where outliers exist, up to five randomly chosen are listed in a table in the Summary Report, whereas the Full Report will list all the outliers found.
It has the following columns:
| Mol | The molecule identifier |
| Chain | The instance identifier |
| Res | The residue number Type: :The residue name |
Side chains of asparagine, glutamine and histidine can sometimes be rotated ("flipped") to make optimal hydrogen bonds, improving its contacts with its neighbours, without affecting their fit to the experimental electron density (see (Chen et al., 2010) for more details).
The sidechain flipping table in the Summary Report lists up to five sidechains for which being "flipped" improves their contacts with atomic neighbours, whereas the Full Report will list all such sidechains. It has the following columns:
| Mol | The molecule identifier |
| Chain | The instance identifier |
| Res | The residue number |
| Type | The residue name |
5.3.3. RNA
This section describes the quality of RNA chains using MolProbity’s analysis of ribose sugar puckers and rotameric nature of "suites" of backbone torsion angles (see Richardson et al., 2008, and Chen et al., 2010 for details). A suite consists of the torsion angles between the sugars in two RNA nucleotides and is identified by the 3' nucleotide.
The summary table summarises the geometrical quality of an RNA chain using the following columns:
| Mol | The molecule identifier |
| Chain | The instance identifier |
| Analysed | The first number here is the number of backbone suites for which analysis was carried out, and the latter number is the total number of nucleotides. The former is a smaller number because a suite is not defined at 5'-end, or a suite might be incompletely modelled. |
| Backbone outliers | The percentage of nucleotide suites in the chain which Molprobitiy identified as an outlier. |
| Pucker outliers | The percentage of sugar pucker outliers in the chain which Molprobitiy identified as an outlier. These are nucleotides where the strong correlation between sugar pucker and distance between the glycosidic bond vector and the following phosphate is violated. |
| Suiteness | The overall suiteness parameter as defined by Molprobity. |
Where backbone or pucker outliers exist, up to five randomly chosen are listed in a table in the Summary Report, whereas the Full Report will list all the outliers found.
Both tables have the following columns:
| Mol | The molecule identifier |
| Chain | The instance identifier |
| Res | The residue number. |
| Type | The residue name. |
5.4 ⇒ 5.7. Non-standard residues in protein, DNA, RNA chains; Carbohydrates; Ligand geometry; Other polymers
These sections analyse the geometry of:
Non-standard amino acids within proteins and non-standard nucleotides within DNA or RNA
Carbohydrates
Ligands
Other polymers
Bond lengths, bond angles, acyclic torsions and isolated rings are assessed using the Mogul program (Bruno et al., 2004) by comparison with preferred molecular geometries derived from high-quality, small-molecule structures in the Cambridge Structural Database (CSD). Chirality is assessed by Validation-pack (Feng et al.).
There are two summary tables providing a per-molecule overview and detailed tables that provide information on (some of) the outliers for each criterion (if any; otherwise the table is omitted).
Summary table for bond lengths and angles
A Z-score is calculated for each bond length and bond angle in the molecule (A Z-score is generally defined as the difference between an observed value and an expected or average value, divided by the standard deviations of the latter.). Individual bond lengths or angles with a Z-score less than -2 or greater than 2 merit inspection.
The root-mean-square value of the Z-scores (RMSZ) of bond lengths (or angles) is calculated for the whole molecule. RMSZ scores are expected to lie between 0 and 1. For low-resolution structures, geometry should be tightly restrained and small values are expected. For very high-resolution structures, values approaching 1 may be attained. Values greater than 1 indicate over-fitting of the data.
At least 20 examples were required for each bond length and bond angle to be assessed.
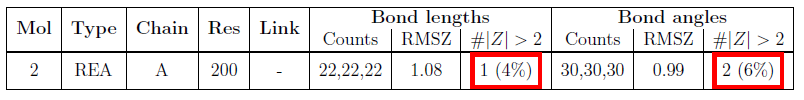
The bond/angle summary table has the following columns:
| Mol | The molecule identifier. |
| Type | The residue name. |
| Chain | The instance identifier. |
| Res | The residue number. |
| Link | The identifier(s) of the molecule(s) to which the residue is linked, e.g. by a covalent bond, salt bridge etc. |
| Bond lengths (or angles) | This column is subdivided into three:
|
Summary table for chirality, torsions and rings
For acyclic torsion angles, Mogul provides the local density measure. This measures the ratio of incidences in the Cambridge Structural Database within 10 degrees of the torsion angle in question, to the number of total incidences of the torsion angles in the Cambridge Structural Database. If this figure was less than 5% the torsion angle is considered an outlier.
For isolated rings, Mogul compares the given ring with comparable rings in small molecules structures in the Cambridge Structural Database and calculates an RMSD value based on corresponding constituent torsion angles for each comparable ring. The mean and minimum of these RMSDs both have to be above 60° for the ring to be flagged an outlier.
At least 15 examples were required for each torsion angle and ring to be assessed.
Note that the criteria used to flag a ring or torsion angle as an outlier are under development. The current criteria are very conservative. They will be refined following analysis of a large test set of ligands.
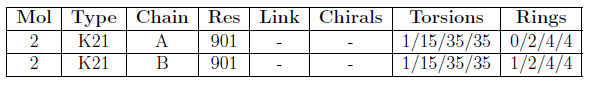
The chirality, torsion angles and rings summary table contains the following columns:
| Mol | The molecule identifier. |
| Type | The residue name. |
| Chain | The instance identifier. |
| Res | The residue number. |
| Link | One or more molecule identifiers to which the residue is linked, e.g. by a covalent bond, salt bridge etc. |
| Chirals | This column lists: the number of chiral outliers in the chain, the number of chiral centers analysed, the number of these observed in coordinates and the number defined in the PDB chemical component dictionary. |
| Torsion | This column lists: the number of torsion angle outliers in the chain, the number of torsions analysed, the number of these observed in coordinates and the number defined in the PDB chemical component dictionary. |
| Rings | This column lists: the number of ring outliers in the chain, the number of rings analysed, the number of these observed in coordinates and the number defined in the PDB chemical component dictionary. |
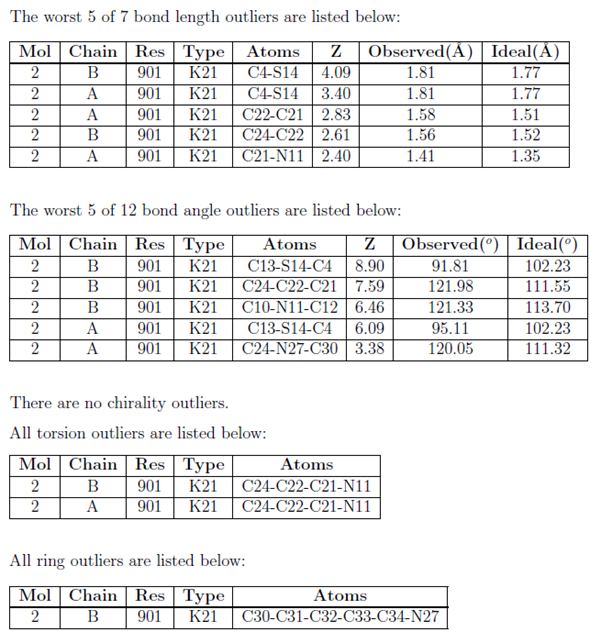
Information tables for bond length, bond angle, chirality, torsion angle and ring outliers
Where outliers exist, up to five for each category are listed in a table in the Summary report, while the Full report lists all of them. Bond length and bond angle outliers are sorted by the Z-score (worst first). Other outliers are selected randomly in the Summary report.
The outlier tables have the following columns in common:
| Mol | The molecule identifier. |
| Type | The residue name. |
| Atom(s) | names of atoms involved in the bond, angle, torsion angle, ring, or the name of the chiral atom with the unusual deviation. |
| Chain | The instance identifier. |
| Res | The residue number. |
The following columns are specific to the bond length and bond angle outliers tables:
| Z | The difference between observed and ideal values in terms of standard deviations. |
| Observed | The observed value of the bond length or angle. |
| Ideal | The ideal value of the bond length or angle. |
For example: |
The two-dimensional graphical depiction (Smart and Bricogne, 2015) of Mogul quality analysis of bond lengths, bond angles, torsion angles, and ring geometry are provided for ligands that have been designated as ligand of interest (LOI) by the depositor, regardless of the validation assessment, and for any ligands with molecular weight greater than 250 Daltons that have outliers flagged in validation.
Color scheme is coded according to validation result with green indicating commonly observed values, magenta indicating unusual values, and gray indicating that there was insufficient data to derive a validation score. Unusual values include model quality and electron density fit. For model quality, individual bond lengths or angles with a Z-score less than -2 or greater than 2, the torsion angle with less than 5% of local density measure from Mogul calculation, or RMSD is above 60 degree are considered unusual and colored in magenta.
6. Fit of model and data
This section presents analysis of the fit of the molecules in the entry to the experimental data. Electron density maps are calculated by Servalcat (command refmacat) (Yamashita et al., 2023) from the coordinates in the entry and the experimental X-ray structure factor data using a procedure adapted from Uppsala Electron-Density Server (Kleywegt et al., 2004). The fit of the model to the 2mFo-DFc electron density map is assessed with Real Space R-value (RSR) computed by the density-fitness program (which was previously called stats) (van Beusekom, Joosten et al., 2018). This analysis is performed on a per residue / nucleotide basis. Lower values of RSR indicate a closer match between the experimental and calculated electron density, and thus a better fit of the residue / nucleotide to the experimental data.
RSRZ is a derived measure which normalises RSR against residue type and resolution. RSRZ values of greater than 2 indicate an outlier. RSRZ is defined only for standard amino acids and nucleotides, due to the lack of sufficient data to make statistically significant conclusions for other molecules (Gore et al., 2017).
6.1. Protein, DNA and RNA chains
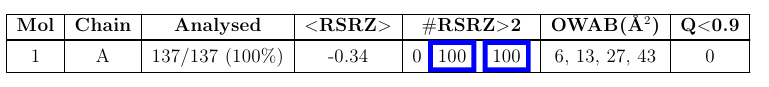
This section describes the fit between the experimental electron density and standard residues / nucleotides in protein, DNA and RNA chains. The first table summarises the quality using the following columns:
| Mol | The molecule identifier |
| Chain | The instance identifier |
| Analysed | This column provides the number of residues in the chain for which RSRZ was analysed over the total number of residues present in the chain. In parentheses this value is expressed as a percentage. |
| <RSRZ> | The mean value of the per-residue RSRZ. |
| #RSRZ > 2 | This column is separated into three sub columns: the number of residues for which RSRZ is is less than -2 or greater than 2. The next two columns compare the number of residues / nucleotides with RSRZ less that -2 or greater than 2 per number of residues / nucleotides analysed relative to all X-ray structures available in the PDB archive prior to 2011 (2nd column) or a subset with similar resolution to this entry. |
| OWAB | The Occupancy-Weighted Average B (OWAB) value per residue (in units Å2). This value is calculated by multiplying the B factor for each atom in the residue by its occupancy and then averaging this value over all atoms in the residue. The OWAB column is then presented as 4 sub columns providing: the minimum, median, 95th percentile and maximum OWAB value for all residues in the instance being analysed. |
| Q < 0.9 | This is defined as the number of residues in the instance which have an average occupancy of less than 0.9. In parentheses this value is expressed as a percentage of total observed residues / nucleotides in the instance. |
If any RSRZ outliers have been identified then the next table will either list all of them (if this is a full report) or the worst 5 (for a summary report).
| For structures with resolutions worse than 3 Šand residues with B-factors above 90 Ų, RSR/RSCC values computed from density-fitness may be unreliable and should be interpreted with caution. |
6.2 ⇒ 6.5. Non-standard residues in protein, DNA, RNA chains; Carbohydrates; Ligands; Other polymers
These sections describe the fit between the experimental electron density and:
Non-standard amino acids with proteins and non-standard nucleotides within DNA or RNA
Carbohydrates
Ligands
Other polymers
For which RSRZ values cannot be calculated due to insufficient instances within the PDB archive for statistically significant conclusions to be drawn (Kleywegt et al., 2004).
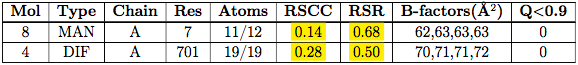
The Non-standard residues, Carbohydrates, Ligands and Other polymer tables present the same information. For example these are results for a ligand with a good fit to electron density:
 ,
,
whereas this is the table for a structure with a ligand DIF whose placement has been questioned (Smart et al., 2018):
 .
.
The columns are labelled:
| Mol | The molecule identifier. |
| Type | The name of the molecule. |
| Chain | The instance identifier. |
| Res | The instance residue number. |
| Atoms | The two numbers in this column represent the number of atoms modelled in the instance over the number of atoms defined for the instance type in the (CCD). |
| RSCC | The real space correlation coefficient for the instance. This is an alternative to RSR for assessing how well the ligand’s calculated electron density map matches the EDS electron density map calculated from the experimental diffraction data. A value above 0.95 normally indicates a very good fit. RSCC around 0.90 are generally OK. A poor fit results in a value around or below 0.80 that may well indicate the experimental data do not accord with the ligand placement. (Smart et al., 2018). |
| RSR | The Real-space R value a measure of how well ‘observed’ and calculated electron densities agree for the ligand. From a user perspective, it is important to note that the range of RSR is from 0 meaning ‘perfect agreement’, with values approaching or above 0.4 indicating a poor fit and/or low data resolution (Smart et al., 2018). |
| B-factors | Four numbers are listed: the minimum, median, 95th percentile and maximum B-factors for all atoms in the instance being analysed. The B-factor is also known as the Debye-Waller factor or "temperature factor" and is a quantity indicating the spread of the atom in the refined model. It units are Å2. A value approaching 80 Å2 indicate the atom is spread very widely and the residue/ligand may be disordered. |
| Q < 0.9 | Lists the number of atoms in the instance which have an occupancy of less than 0.9. |
| ligands whose RSCC value is below 0.8 and RSR value is less than 0.4 are highlighted in yellow because this an indication of a poor fit to electron density. |
| In the past a density fit metric called LLDF was used to assess ligand electron density fit but this has been dropped because it was found to give misleading results in some cases (Smart et al., 2018). |
The graphical depiction of the model fit to experimental electron density (Smart and Bricogne, 2015) are provided for ligands that have been designated as ligand of interest (LOI) by the depositor, regardless of the validation assessment, and for any ligands with molecular weight greater than 250 Daltons that have outliers flagged in validation. Outliers include model quality and electron density fit which is calculated RSCC value below 0.8 and calculated RSR value less than 0.4.
For the color scheme, the 2mFo-DFc density map is shown in gray, while the positive and negative mFo-DFc difference density maps are shown in green and magenta, respectively.
2024年11月12日 | カテゴリー:AUTODOCK VINA , 創薬/AUTODOCK |