p16遺伝子について
サイクリン依存性キナーゼ阻害2A (cyclin-dependent kinase inhibitor 2A) は、CDKN2Aとも言い、ヒトのCDKN2A(p16)遺伝子によりコード化されているがん抑制タンパク質である[5][6][7]。p16は細胞周期の調整に重要な役割を果たしており、p16の変異は様々な癌、特にメラノーマの発生のリスクを高めている。なお、CDK(cyclin-dependent kinase)とはサイクリン依存性キナーゼの略である。
機能
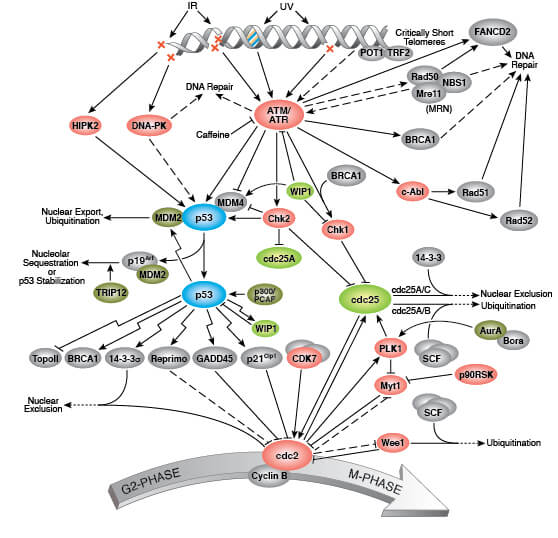
p16遺伝子は、最初のエクソンが異なるいくつかの転写変異体を生成する。明確なタンパク質をコード化している少なくとも3つの可変な結合変異体が報告されており、そのうち2つはCDK4キナーゼ阻害機能として知られている構造的に関連した異性体をコード化している。遺伝子の残りの20000塩基対分の上流に位置する最初のエクソンを含んだ残りの転写変異体は、腫瘍抑制タンパク質p14ARF(ARF)を含んでいる。このARFは癌抑制タンパク質であるp53の働きを弱める作用を有するMDM2に干渉し隔離するため、ARFはP53を安定化する役割を持っている[8]。
構造的及び機能的な相違に関わらず、細胞周期のG1期のCDK4及びp53の調整機能を通じてp16遺伝子によりコード化されたCKD阻害異性体及びAFR生成物が細胞周期のG1期で通常の機能の役割を行っている。p16遺伝子は、様々な癌においてしばしば変異しているか欠落しており、重要ながん抑制遺伝子として知られている[5]。
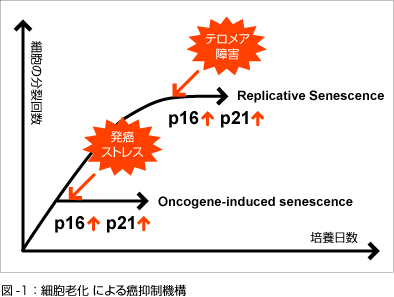
組織が老化したときのp16遺伝子の発現の増加は幹細胞の増殖を減退させる[9]。細胞老化と関連して癌のリスクは増加しているが、幹細胞の分裂と増殖のこれらの減退は、癌からの脅威から守っている。
臨床的意義
CDKN2A(p16)遺伝子の変異は、幅広い癌の発生のリスクに関連しており、遺伝子の変異は癌の培養細胞で多く認められている[10][11]。例えば次のような例がある。
- 膵臓腺癌は、CDKN2A(p16)遺伝子の変異としばしば関係している[12][13][14]。
- p16の欠落は、食道癌と胃癌の培養細胞からしばしば見つかっている[15]。
- p16INK4aの濃度は、組織の老化に伴って飛躍的に上昇する。それゆえp16INK4aは、分子レベルでどの程度早く組織が老化しているかを測定する血液検査に応用できる可能性がある[15]。
- p16蛋白に対する免疫染色はHPV検査に対する病理組織上での代理マーカーである。HPVに感染して宿主細胞の細胞増殖機構に異常をきたした不死化細胞はp16過剰発現を示す。[16]

Cellular senescence is thought to contribute to age-associated deterioration of tissue physiology. The senescence effector p16(Ink4a) is expressed in pancreatic beta cells during aging and limits their proliferative potential; however, its effects on beta cell function are poorly characterized. We found that beta cell-specific activation of p16(Ink4a) in transgenic mice enhances glucose-stimulated insulin secretion (GSIS). In mice with diabetes, this leads to improved glucose homeostasis, providing an unexpected functional benefit. Expression of p16(Ink4a) in beta cells induces hallmarks of senescence--including cell enlargement, and greater glucose uptake and mitochondrial activity--which promote increased insulin secretion. GSIS increases during the normal aging of mice and is driven by elevated p16(Ink4a) activity. We found that islets from human adults contain p16(Ink4a)-expressing senescent beta cells and that senescence induced by p16(Ink4a) in a human beta cell line increases insulin secretion in a manner dependent, in part, on the activity of the mechanistic target of rapamycin (mTOR) and the peroxisome proliferator-activated receptor (PPAR)-γ proteins. Our findings reveal a novel role for p16(Ink4a) and cellular senescence in promoting insulin secretion by beta cells and in regulating normal functional tissue maturation with age.
Ageing is generally associated with deterioration of organ function and regenerative potential. In the case of pancreatic β-cells, an age-related decline in proliferative potential is well documented, and was proposed to contribute to the increased prevalence of type 2 diabetes in the elderly. The effects of ageing on β-cell function, namely glucose-stimulated insulin secretion (GSIS), have not been studied as extensively. Recent work revealed that, surprisingly, β-cells of mature mice and humans secrete more insulin than young β-cells in response to high glucose concentrations, potentially serving to counteract age-related peripheral insulin resistance. This functional change appears to be orchestrated by p16(Ink4A) -driven cellular senescence and downstream remodelling of chromatin structure and DNA methylation, enhancing the expression of genes controlling β-cell function. We propose that activation of the cellular senescence program drives life-long functional maturation of β-cells, due to β-cell hypertrophy, enhanced glucose uptake and more efficient mitochondrial metabolism, in parallel to locking these cells in a non-replicative state. We speculate that the beneficial aspects of this process can be harnessed to enhance GSIS. Other age-related mechanisms, which are currently poorly understood, act to increase basal insulin secretion levels also in low glucose conditions. This leads to an overall reduction in the amplitude of insulin secretion between low and high glucose at old age, which may contribute to a deterioration in metabolic control.
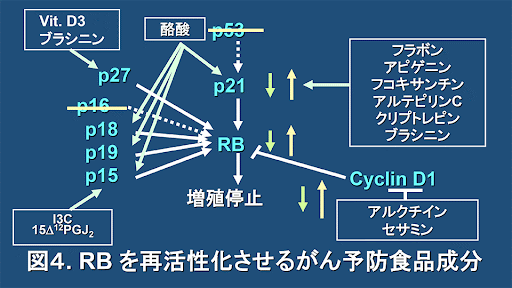
p16Ink4a (p16) is an important tumor suppressor which is upregulated in senescent cells and in aged tissues. p16 acts as an inhibitor of the interaction between Cyclin-Dependent Kinases (CDK) 4/6 and CyclinD1 leading to the activation of retinoblastoma protein (RB). Consequently, active RB interferes with the translocation of E2F1 into the nucleus and arrests cells in the G1-S phase of the cell cycle [1].
In cancer cells with mutations in RB or CDK4/6, p16 is normally overexpressed but unable to induce cell cycle arrest. p16 -overexpressing cancer cells are found in different types of carcinomas and are considered highly aggressive and invasive [2].
Several drugs in recent years have been shown to have ‘senolytic’ properties (i.e. being toxic for senescent cells) and to remove p16+ cells from a variety of tissues [3]. Among these compounds, ABT-737 and its orally-available analogue ABT-263 target the anti-apoptotic proteins BCL-2, BCL-W and BCL-XL, considered essential pro-survival players in senescent cells. The effects of these compounds in mice almost completely overlap with a suicide gene strategy activated by the p16 promoter, thus suggesting specific targeting p16+ cells [3]. However, when we tested ABT-737 and ABT-263 against p16-overxpressing murine sarcomas we failed to observe any toxicity, despite p16+-cancer cells upregulating both BCL-2 and BCL-XL [4]. These data could be interpreted in 3 ways: 1) ABT compounds are specifically active against non-proliferating p16+-cells; 2) the efficacy of ABTs requires upregulation of BCL-W, which we have recently shown being a common feature of senescent cells [5]; 3) ABTs act independently of p16 expression levels. The latter hypothesis would represent a critical issue, as p16 is used as a major readout for the efficacy of senotherapies.
Since we have recently developed an inducible suicide gene regulated by the full p16 promoter [6], we have then studied whether the use of this strategy could be effective against p16+ tumors. Indeed, most p16 -overexpressing cancer cells were efficiently eliminated by the activation of the suicide gene in both culture and in vivo conditions [4]. These data suggest that p16 upregulation is maintained by active transcription, possibly mediated by emergency signaling pathways attempting to restrain cellular proliferation.
Our study supports the idea that the overexpression of oncosuppressors could be exploited for interventions against cancer. While we studied a specific context in which p16 is present in its wild-type form, this strategy could potentially work in situations of overexpression (by transcriptional regulation) of mutated forms, which is a common feature of cancer cells. On this line, similar strategies against additional oncosuppressors such as p14 and p53 could be effective.
In parallel, it will be of interest to understand whether a p16 -based suicide gene therapy could be used in other contexts. Studies in transgenic mouse models have shown that elimination of p16+ cells using suicide genes can significantly delay the onset and progression of a number of age-related pathologies, eventually leading to lifespan extension [7]. Whether a similar strategy could be used for human interventions is still matter of debate. Despite significant efforts and resources have been spent in the last two decades for the development of suicide gene therapies, particularly for cancer treatment, none of these therapies have been approved by regulatory agencies. Indeed, there are still a number of major limitations preventing the feasibility of suicide gene strategies for human treatment. First, damaged or mutated cells responsible to promote onset and progression of disease might be heterogeneous and might not express the same markers, thus limiting the efficacy of a therapy based on interfering with specific genes. Second, current suicidal strategies might not be entirely safe. For example, one of the most used suicide gene is the herpex simplex virus thymidine kinase (HSV-tk), shown to have significant bystander effects. Third, the delivery methods for genes in vivo are highly inefficient and not yet entirely safe to use in humans.
The advent of technical and intellectual breakthroughs overcoming these limitations could potentially resolve in an innovative, accessible and adaptable technology for the cure of many diseases.
2024年10月29日 | カテゴリー:癌の病態生理と治療学 |