
大腸癌細胞の増殖シグナル
大腸がん細胞の増殖に関与する有名なシグナル伝達経路には、以下のものがあります:
- Wntシグナル経路: 大多数の大腸がんでは、この経路が異常に活性化しており、特に癌遺伝子c-Mycの異常発現が重要な役割を果たしています1。Wnt シグナル(注1)の異常な活性化が大腸癌発症の最も大きな原因であると考えられて
います。Wnt シグナルはたくさんの遺伝子の発現を引き起こしますが、その中でも転写因子を
コードする c-Myc 遺伝子(注2)はがんの発症に最も重要な因子であると考えられています。
しかしながら、Wnt/c-Myc ががんの発症を誘導する仕組みについては未だ不明な点が残されて
います。東京大学分子細胞生物学研究所の秋山教授、川崎准教授らの研究グループは、ほとん
どの大腸がんで Wnt/c-Myc がタンパク質をコードしない新規の長い RNA〝MYU〞の発現を
誘導していることを見いだしました。さらに、①MYU は大腸がん細胞が腫瘍をつくるために
必須の役割を果たしていること、②MYU は細胞周期を進める機能をもつタンパク質 CDK6(注
3)の発現亢進を引き起こしていること、③MYU による CDK6 の発現亢進が大腸がん細胞の
増殖の大きな要因であることを見出しました。大腸がんの新しい治療戦略として、 MYU およ
び MYU が腫瘍をつくる仕組みを標的とした薬剤の創製が期待されます。 - Ras/Raf/MEK/ERK (MAPK)経路: この経路は細胞増殖、成長、分化などを制御し、がん細胞の増殖シグナル伝達において重要です2。
マイトジェン活性化プロテインキナーゼ(MAPK)カスケードは、細胞増殖、成長、分化、形質転換およびアポトーシスなどの多種多様な細胞プロセスを制御する複雑なシグナル伝達ネットワークを形成します。MAPK経路のキーメディエーターの調節不全は、腫瘍形成のドライバーとして関与し、がん細胞に以下の能力-マイトジェンシグナルからの独立、増殖シグナル伝達の維持、アポトーシス回避能力、抗増殖シグナルに対する感受性、転移能、および血管新生能-を付与します。MAPK経路の変異が腫瘍促進プロセスに寄与しているという仮定のもと、本経路の阻害剤を開発するための研究に多くの力が注がれています。
主要なMAPK経路の構築
マイトジェン活性化プロテインキナーゼ(MAPK)シグナル伝達のノードは、酵母からヒトにいたる真核生物における増殖、細胞分裂、代謝、運動性、自然免疫、細胞ストレス反応、アポトーシス、および生存機能を含む極めて重要な細胞機能を制御、微調整するための細胞外刺激を伝達し、調節します。MAPK経路には、4種類の主要な分岐経路、および十数種類のMAPK酵素の存在が知られており、少なくとも7種類の異なるグループに分類されます(図4.1)*1–3。
ERK経路は典型的に増殖シグナル伝達に関与する増殖因子によって誘導されるのに対し、JNK、p38、およびERK5経路は主にアポトーシス、増殖阻害、自然免疫、および細胞ストレス反応関連の刺激によって活性化します。これらのそれぞれの古典的MAPKカスケードは、二重特異性セリン/スレオニンプロテインキナーゼ(MAPK、MAPK活性化因子(MEK、MKK、またはMAPK キナーゼ)、ならびにMEK活性化因子(MEKキナーゼ [MEKK]またはMAPKキナーゼキナーゼ)を介する3種類のプロテインキナーゼによる連続的な活性化を特徴とします。古典的MAPK経路の活性化は細胞膜において開始され、そこで低分子量GTPアーゼおよびさまざまなプロテインキナーゼによってMAPKKKがリン酸化されることで活性化されます。続いて、MAPKKKによってMAPKKが直接リン酸化され、活性化されたMAPKKはMAPKをリン酸化します。活性化されたMAPKは、多数の細胞質内基質と相互作用してリン酸化を行い、最終的に状況特異的な遺伝子発現を誘導する転写因子を調節します。この結果、次々に多様な生物学的反応、例えば、浸透圧ショック、細胞周期進行、あるいはインターフェロン産生の誘導が生じます *3,4。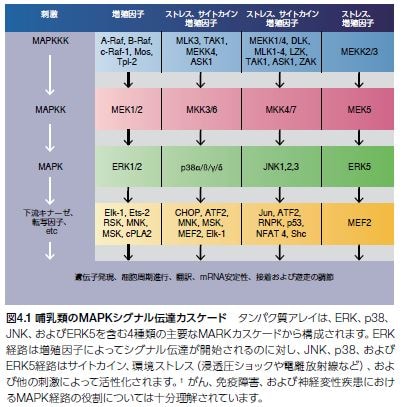
MAPK経路の複雑性
MAPKシグナル伝達カスケードは、シンプルな直線的で一方向性のプロテインキナーゼ群として表されますが、その経路は非常に複雑です。MAPKカスケードおよび他のシグナル伝達ネットワークの中には、程度の大きいクロストークが存在します。例えば、MAPK、PI3Kネットワーク、NFκB、およびJAK-STAT経路のメディエーター間の相互作用については十分に立証されています*1,7。

当記事は、がん増殖シグナル伝達経路の探索ガイドブックからの抜粋です。
- PI3K/Akt/mTOR経路: この経路も細胞の増殖や生存に関与し、がん細胞の増殖を促進します2。
キナーゼ(MAPK)カスケードは、細胞増殖、成長、分化、形質転換およびアポトーシスなどの多種多様な細胞プロセスを制御する複雑なシグナル伝達ネットワークを形成します。MAPK経路のキーメディエーターの調節不全は、腫瘍形成のドライバーとして関与し、がん細胞に以下の能力-マイトジェンシグナルからの独立、増殖シグナル伝達の維持、アポトーシス回避能力、抗増殖シグナルに対する感受性、転移能、および血管新生能-を付与します。MAPK経路の変異が腫瘍促進プロセスに寄与しているという仮定のもと、本経路の阻害剤を開発するための研究に多くの力が注がれています。
Rasタンパク質は、HRAS、KRASおよびNRASの3種類のアイソタイプが発見されており、細胞周期進行、細胞移動、アポトーシス、老化、および他の生体機能に関与する多くのシグナル伝達経路に関連する低分子量グアノシントリホスファターゼ(GTPアーゼ)です。Ras活性化の簡単な説明図を図4.3に示します。*8,9–10 SRCは最初のがん遺伝子として同定されていますが、Rasは最初のヒトがん遺伝子として同定されています。また、Rasファミリーメンバーにおける変異はヒト腫瘍の約30%において検出されています*11–13。
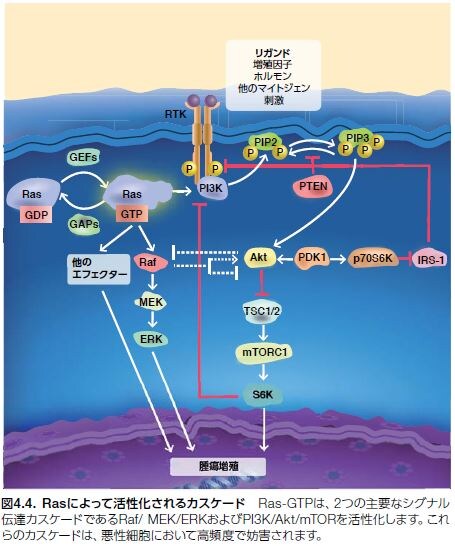
多数の細胞表面分子によりRasタンパク質が活性化されると、がんにおける役割が明確となっているRasの主要なエフェクター経路であるRaf/MEK/ERKなどのMAPKカスケードが次々に活性化されます。PI3K/Akt/mTOR経路は、Ras依存性およびRas非依存性のメカニズムによって活性化され、増殖因子およびGタンパク質共役受容体のMAPK経路下流と収束点を共有します*11 。シグナル伝達イベントは、Raf、MEK、およびERK1/2(MAPK1/3とも呼ばれる)を介して導入されるRasによって開始され、核まで伝達されます。そこで、FOS、MYC、ELK、および c-JUNなどの転写因子が制御されることで、細胞増殖および生存に必要な遺伝子発現が調節されます。(図4.2および図4.4)*6,11,14。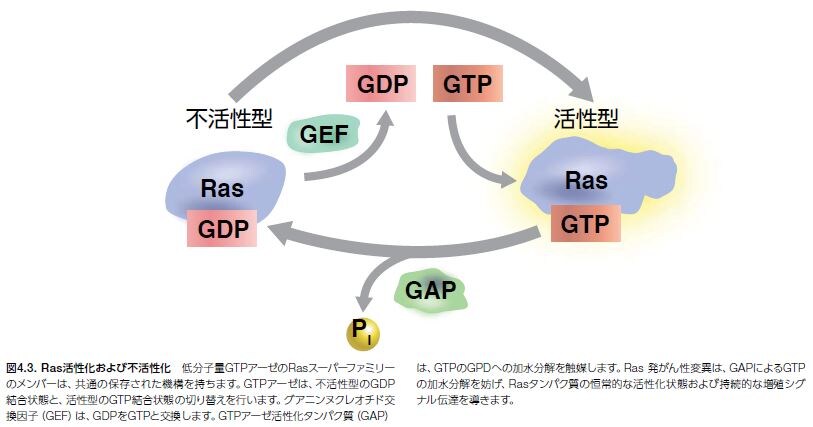
がんにおけるRas/Raf/MEK/ERK経路の役割
Ras活性化のエフェクター
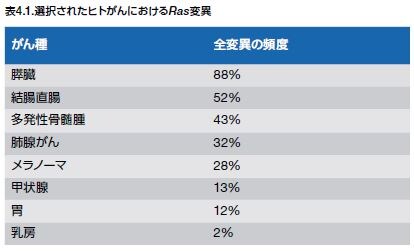
Ras変異は、血液腫瘍および固形腫瘍の両方において同定されています。NRASは、血液腫瘍およびメラノーマにおいて最も高頻度で変異が見られるアイソフォームであるのに対し、KRASは、結腸がんおよび肺がんにおいて高頻度で変異が見られます。完全には網羅していませんが、Ras変異を持つことが知られているヒトがんのリストを表4.1に示します。アイソフォーム特異的なRas変異の頻度および他のMAPK経路の遺伝子を図4.5に示します。これまでにRas阻害剤は開発されていませんが、下流のRas/Raf/MEK/ERKメディエーターについてはいくつかの阻害剤を開発することに成功しています *15–17 。
Ras/Raf/MEK/ERKシグナル伝達の阻害剤
Rasシグナル伝達の下流エフェクターを阻害する薬剤が開発されつつあります。現在、3種類のRas/Raf/MEK/ERK経路阻害剤が医薬品規制機関に承認されています。2種類はBRAFを標的としますが、このMAPKKKは活性型Rasタンパク質の直接的な基質として働きます。3番目の阻害剤は、BRAFの下流で機能するMEKを標的とします(図4.6および図4.7)。
BRAFがん原遺伝子は、ARAF、BRAF、およびCRAF(RAF1とも呼ばれる)の3種類のアイソフォームが知られるRafファミリーメンバーをコードします。これらの高度に保存されたセリン/スレオニンキナーゼは、MAPK [Ras/Raf/MEK/ERK] 経路のメディエーターです。Rafタンパク質は、上流キナーゼRasの基質、およびMEKの直接的な活性化因子として働きます。Rafの過剰活性化は増殖、分化、およびアポトーシスの調節不全につながり、発がん性Ras変異は種々のヒトがんにおいて同定されています。BRAFアイソフォームの変異は、悪性腫瘍̶特に、メラノーマならびに甲状腺がんおよび結腸がんにおいて最も高頻度に認められますが、ARAFやCRASにも発がん性変異は生じます *18。
2002年、一塩基のミスセンス置換(1,799位のヌクレオチドにおけるT→A)によって、BRAFのコドン600においてバリンがグルタミン酸に置換される(V600E)ことが研究者により報告されました。全BRAF変異の80%は高い発がん性を示します。BRAF変異の残りの20%は、V600の近くに位置するエキソンにおいてミスセンス変異が生じています。これらのBRAF変異は、BRAFキナーゼ活性を損なわせる、MAPK経路の持続的な活性化因子です(図4.5)*18,19。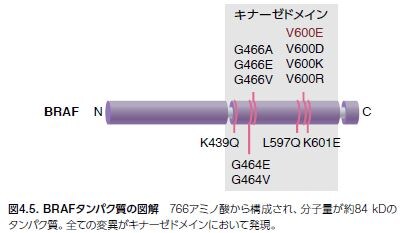
BRAF阻害剤
RasおよびBRAFの変異はいずれも強い発がん性を示し、正常細胞における悪性形質転換を誘導することで知られています。Rasの低分子阻害剤については、これまでの臨床研究で進展がみられていませんが、2種類のBRAF阻害剤―ベムラフェニブおよびダブラフェニブ―については、現在、特定の型のメラノーマ患者を対象とした使用が可能となっています(図4.6)。
ベムラフェニブは2011年に、続いてダブラフェニブは2013年に、V600E変異を有する切除不能または転移性メラノーマに対する治療薬として承認されました。いずれのBRAF阻害剤も、野生型BRAFを有するメラノーマの治療には使用されません *20–23。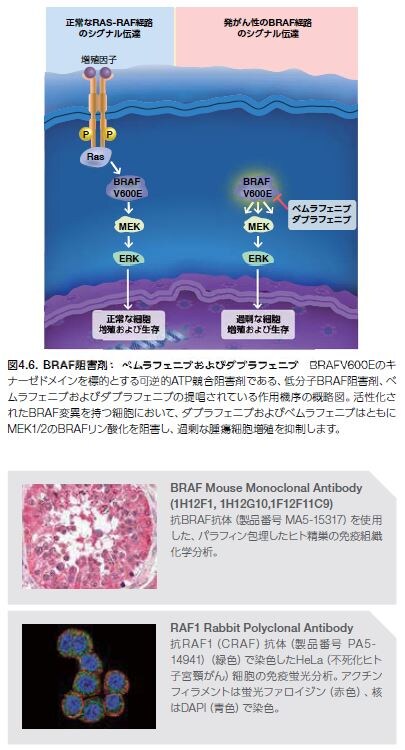
MEK阻害
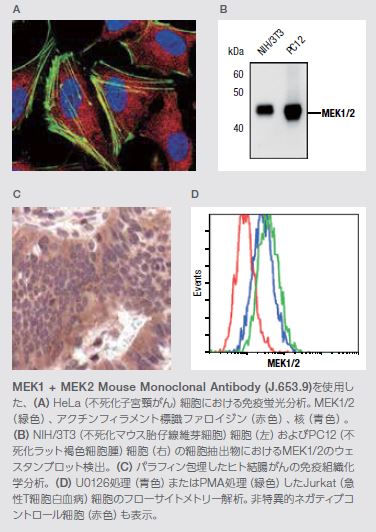
MAPKKs、MEK1およびMEK2は、ERK1およびERK2上のセリン残基およびスレオニン残基を選択的にリン酸化するキナーゼであり、Ras/Raf/MEK/ERKシグナル伝達カスケードの末端キナーゼです。いったん活性化されると、ERK1およびERK2は、細胞周期進行、分化、運動性、代謝、および血管新生などの幅広い細胞プロセスを調節している複数の基質―核および細胞質のリン酸化を触媒します。MEK1およびMEK2キナーゼ遺伝子に遺伝子変異が生じることは稀ですが、原発ヒト腫瘍の解析においてMEK活性の上方制御が頻繁に認められています。トラメチニブは、V600EまたはV600K BRAF変異陽性のメラノーマに対する阻害活性が示されているMEK阻害剤です(図4.7)*16,24–25。
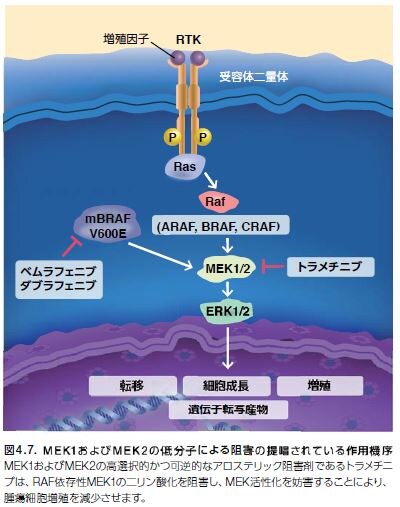
MEK1/2は、マイトジェン活性化プロテイン(MAP)キナーゼキナーゼとして機能する二重特異性プロテインキナーゼファミリーのメンバーです。MAPキナーゼは、細胞外シグナル制御キナーゼ(ERK)としても知られ、複数の生化学的シグナルの統合ポイントとして作用します。これらのキナーゼは、MAPキナーゼシグナル伝達経路の必要不可欠な要素として、増殖、分化、転写調節、および発生などの多くの細胞プロセスに関与しています。ERK阻害
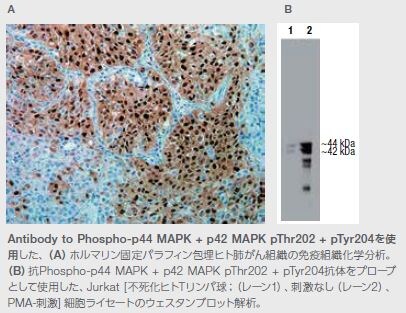
MAPK ERK1およびERK 2(MAPK3およびMAPK1とも呼ばれる)は、G0/G1期からS期までの細胞周期進行を誘導するセリンおよびスレオニンキナーゼです(図4.8)。ERK1およびERK2(ERK1/2)シグナル伝達は、正の細胞周期制御因子であるサイクリンD1およびc-MYCの活性を調節し、Tob1、FOXO3a、およびp21などの負の制御因子を下方制御します。ERK1/2 MAPキナーゼ経路は、細胞分裂周期における直接的な役割の他に、さまざまな機構によって細胞成長および増殖を制御します。すなわち、ERK1/2は、細胞周期メディエーターを直接調節、タンパク質およびヌクレオチドの合成を促進し、TSC2(ツベルンとも呼ばれる)などのタンパク質との相互作用を介してmTORシグナル伝達を上方制御します *26。
さまざまながん細胞株におけるERK1/2シグナル伝達の低分子による阻害は、ERK1/2の封鎖が細胞周期停止を引き起こし、アポトーシスを誘導することを示します。マウス異種移植片モデルを用いたIn vivo薬理試験においても、MEK1/2低分子阻害剤が腫瘍成長の阻害を促進することが示されています。ERK1/2がRas/Raf/MEK/ERK経路の調節およびさまざまながんモデルにおける着実な前臨床試験結果に重要な役割を果たしているとすると、ERK1/2キナーゼを標的とする薬剤の開発を追求するための強い理論的根拠が存在すると言えます。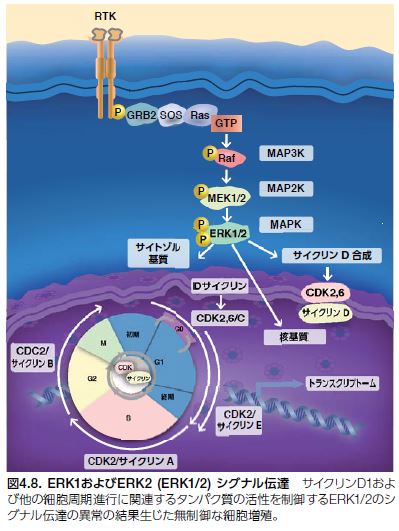 ERK阻害剤については、さまざまながんに対する単独および併用療法について臨床試験が進められています *27–29。
ERK阻害剤については、さまざまながんに対する単独および併用療法について臨床試験が進められています *27–29。
ソラフェニブは、医薬品規制機関による承認済みのERK1/2キナーゼに対する活性を示すRAF阻害剤で、腎臓や肝臓のがん治療に使用されます。しかしながら、ソラフェニブは、複数標的のチロシンキナーゼ阻害剤であるため、ERK1/2活性を下方制御する他に、以下の受容体̶VEGFR、PDGFR、FLT3、RET、およびc-Kitも標的とします(図4.9)*30 。ソラフェニブは数種の異なるタンパク質を標的とするため、いずれのタンパク質を標的としたERK1/2阻害がどの程度ソラフェニブの有効性に寄与しているかは明らかになっていません。選択的ERK阻害剤は開発努力が続けられています *31,32。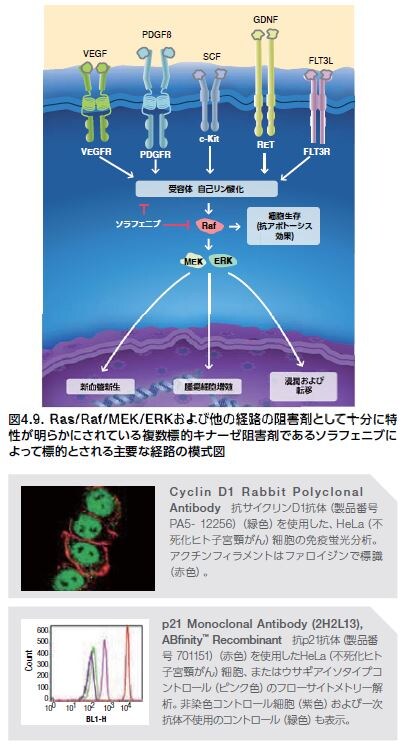
これらの経路は、がん治療のターゲットとしても研究されています。
2024年9月21日 | カテゴリー:各種治療学, 各種病因学, 癌の病態生理と治療学 |


{kind=link}
{kind=link}